
Diumenge se celebra La Marató de TV3, enguany al voltant de les malalties minoritàries. Des de La Fura ens hi sumem dedicant el Tema de la Setmana a explicar què són i a conèixer alguns testimonis de les nostres comarques.
A les malalties minoritàries se les anomena també rares perquè afecten un petit percentatge de la població general (entre un 5% i un 7%). Però si aquest percentatge el traslladem a la població de l’Alt Penedès, Baix Penedès i Garraf ens adonem que entre 18.000 i 24.000 persones en pateixen alguna.
Aquest tipus de malalties són greus, poc freqüents i amb una afectació màxima de 5 casos per cada 10.000 pe rsones. Més del 80% d’elles tenen una base genètica, és a dir, que la causa és l’alteració d’un gen, i normalment és heretada dels progenitors. Per això, moltes també són hereditàries. Aquest tipus de malalties afecten diversos òrgans i sistemes del cos i provoquen una discapacitat intel·lectual o física. Actualment hi ha més de 7.000 malalties rares identificades que poden afectar de formes molt diferents la salut de les persones malaltes, com ara la mobilitat, el sistema nerviós o l’immunològic, el metabolisme o l’equilibri hormonal, entre altres.
rsones. Més del 80% d’elles tenen una base genètica, és a dir, que la causa és l’alteració d’un gen, i normalment és heretada dels progenitors. Per això, moltes també són hereditàries. Aquest tipus de malalties afecten diversos òrgans i sistemes del cos i provoquen una discapacitat intel·lectual o física. Actualment hi ha més de 7.000 malalties rares identificades que poden afectar de formes molt diferents la salut de les persones malaltes, com ara la mobilitat, el sistema nerviós o l’immunològic, el metabolisme o l’equilibri hormonal, entre altres.
Són malalties cròniques i en molts casos també degeneratives que tenen una gran afectació en la vida diària dels pacients i les seves famílies. Poden fer-se visibles des del naixement o la infantesa, encara que aproximadament la meitat no apareixen fins a l’edat adulta.
1. La paradoxa de la raresa

Com hem indicat, se’n diuen minoritàries perquè afecten un percentatge baix de la població, però quan parlem de xifres absolutes la dimensió és molt més important: a Catalunya afecten entre 400.000 i 500.000 persones, a l’Estat espanyol, 3 milions, i a la Unió Europea, més de 30. Per tant, encara que cadascuna de les 7.000 malalties minoritàries, de forma aïllada, sigui molt poc freqüent, totes juntes suposen una gran quantitat de persones afectades. És el que es coneix com la paradoxa de la raresa.
2. La tardança en el diagnòstic

Com que són tan desconegudes, poc freqüents i poc nombroses, de vegades es triga molt temps a diagnosticar-les correctament. Es calcula que les persones afectades per una malaltia minoritària triguen cinc anys de mitjana a rebre un diagnòstic. El pelegrinatge dels malalts i les seves famílies acostuma a començar per l’atenció primària, derivats d’especialista a especialista, fins que finalment arriben a un centre de referència on se’ls fan les proves definitives i s’identifica la malaltia.
Per intentar pal·liar aquesta situació, la Unió Europea, i també Catalunya, ha impulsat la creació de xarxes d’hospitals per intercanviar informació i accelerar-ne el diagnòstic. El seu compromís és que, el 2027, aquestes malalties es puguin diagnosticar en un màxim d’un any.
La comunitat científica apunta que durant tots els anys que no tenen diagnòstic, les persones afectades no poden fer un tractament adequat. Però fins i tot quan ja el tenen, en molts casos no hi ha un tractament disponible. Encara avui hi ha un 40% de malalties minoritàries que no tenen tractament. Són la causa del 35% de les morts de nens menors d’un any i del 50% de les morts d’abans dels 30. Per tant, un tractament adequat pot millorar la qualitat i l’esperança de vida de les persones afectades.
3. Esperança en la investigació

Segons els experts, és important poder detectar les malalties minoritàries en el moment de néixer per poder aplicar de seguida el tractament adequat. És per això que actualment a tots els centres maternals de Catalunya es fa un test neonatal als nadons que inclou 24 malalties minoritàries. Si el test detecta alguna d’aquestes patologies en l’infant, els equips mèdics es posen en marxa per aplicar el tractament que pugui curar-les o frenar-ne el desenvolupament.
En tractar-se de malalties amb pocs pacients, aconseguir fons per a la recerca és més difícil. La majoria dels medicaments que es fan servir per tractar les malalties minoritàries s’anomenen medicaments orfes. Se’n diuen així perquè, com que cadascuna aquestes patologies afecten molt poques persones, el cost d’aquests medicaments és molt alt i el seu finançament, de vegades, difícil.
Actualment la investigació està centrada sobretot en la millora de la capacitat diagnòstica d’unes malalties que en molts casos són difícils de diagnosticar, i en el desenvolupament de noves teràpies. En aquest sentit, s’està avançant molt en teràpies gèniques que consisteixen, a grans trets, en introduir gens funcionals a les cèl·lules per substituir els que funcionen malament i que provoquen la malaltia.
4. Un dia rar
L’any 2008 es va establir que el 29 de febrer seria el Dia Mundial de les Malalties Rares. És un dia també rar, ja que només existeix cada quatre anys, però els altres tres se celebra el dia 28. La iniciativa serveix per visibilitzar aquestes malalties, sensibilitzar la societat sobre la seva existència, explicar les mancances de tractament que reben moltes d’aquestes dolences i, a la vegada, reivindicar més fons per a la investigació i la millora de l’accés a recursos socials i sanitaris.
5. “La teràpia gènica està avançant molt de pressa i creiem que és la solució a moltes d’aquestes mutacions”

Entrevista a Antoni Riera-Mestre, metge internista de l’Hospital Universitari Bellvitge i membre del consell assessor científic de La Marató de TV3
Totes les malalties minoritàries són d’origen genètic?
Més del 90% sí, estan provocades per una mutació en
un gen que fa que una proteïna o enzim no funcioni bé.
I es manifesten més en l’edat pediàtrica que en l’adulta?
Tot i que moltes sí que es manifesten ja en edat pediàtrica, gairebé la meitat de les malalties minoritàries o bé debuten a l’edat adulta o bé no es diagnostiquen fins llavors.
La resta de malalties minoritàries no genètiques, què les provoca?
Són variades, n’hi ha de causa desconeguda, com les malalties autoimmunes o les vasculitis.
Parlem de malalties minoritàries, però el gruix d’afectats per totes elles no és minoritari…
Efectivament. Hi ha unes 7.000 malalties minoritàries que, malgrat que tinguin una baixa prevalença (menys d’un de cada 2.000 habitants), afecten un 5-10% de la població.
Si això ho apliquem a Catalunya, significa que vora mig milió les persones tenen una malaltiaminoritària. A més, la repercussió en el seu entorn fa que afectin molta gent: malalts, familiars, personal mèdic que hi treballa, associacions de pacients…
Per què costa tant diagnosticar una malaltia minoritària?
Hi ha tantes malalties minoritàries que és molt difícil conèixer-les totes. Des que es detecten els primers símptomes fins que es té un diagnòstic, poden passar entre 4 i 6 anys.
Tant de temps?
Malauradament, és una realitat. Sense que sigui en absolut cap crítica cap a l’atenció primària perquè, insisteixo, és impossible conèixer totes les malalties, quan un pacient acudeix al seu centre d’atenció primària amb algun símptoma que no se sap diagnosticar, sol començar un pelegrinatge per diferents especialistes fins que finalment arriba a un hospital que disposi d’una unitat de referència d’aquella malaltia en concret.
I no es pot fer res per escurçar aquest pelegrinatge?
Ja es treballa per escurçar aquest temps de diagnòstic i per oferir-los l’atenció que requereixen. La Marató serà una oportunitat per aconseguir fons per a la recerca, però també per destinar recursos a escurçar el calvari de molts pacients i les seves famílies fins arribar al diagnòstic. Es poden fer treballs en xarxa, més connexions amb la primària, més difusió de les unitats que tenen els hospitals de referència per facilitar a l’atenció primària la tasca de derivar els seus pacients, etc. Cal que entre tots ho posem fàcil.
Què es fa per tractar aquest tipus de malalties?
Moltes vegades aquestes malalties no tenen un tractament curatiu, per això el que es fa són tractaments substitutius, és a dir, donar aquella proteïna o enzim que funciona malament, d’aquesta manera es poden evitar complicacions. És important tractar-les amb el màxim de precocitat possible. Per això cal que el diagnòstic sigui precoç, perquè ens permet avançar-nos i preveure possibles complicacions. Per altra banda, i com que la majoria d’aquestes mutacions són genètiques, el que es fa en aquests casos és també un estudi de cosegregació familiar per veure quins tenen la mateixa mutació que la persona afectada.
Es poden arribar a preveure aquestes malalties?
Prevenció entesa com evitar que apareguin, no. El que sí que es fa és el diagnòstic preimplantacional, és a dir, agafar espermatozous o òvuls (depenent de quin dels dos progenitors és portador de la malaltia), seleccionar aquells que no tenen la mutació i fer una fecundació in vitro per assegurar que el fill no tingui la malaltia.
L’altre camp en el qual s’està treballant i avançant molt és a través de les teràpies genètiques. Hi ha diferents tècniques: una, per exemple, consisteix a introduir
al pacient un virus amb la seqüència correcta del gen que té la mutació i que es barregi amb el seu ADN, amb la qual cosa s’aconsegueix que la seqüència correcta del gen entri al seu ADN i se solucioni la mutació. Això ja s’està fent, més a nivell de recerca, i la teràpia gènica està avançant molt de pressa i creiem que serà la solució a moltes d’aquestes malalties.
La investigació és la clau.
És imprescindible. És la manera d’avançar i trobar solucions a aquestes malalties. La recerca és complicada, implica tenir personal per fer-la, els reactius que s’empren són cars… Ara bé, a Catalunya hi ha molt nivell, es fa molta recerca i la Marató de TV3 hi ajudarà, sens dubte.
A nivell familiar, les malalties minoritàries tenen més afectació que la resta?L’impacte socioemocional que té el diagnòstic d’una malaltia minoritària també és un tret diferencial respecte a altres malalties. Quan expliques a uns pares que el seu fill té una malaltia minoritària, el curs que tindrà, que és genètica, etc., es genera molta angoixa i depressió. De fet a l’Hospital Universitari de Bellvitge vam comparar l’impacte que tenia aquest diagnòstic amb el de càncer i vam observar que genera la mateixa depressió, però més angoixa que en els pacients amb càncer. Per això a les unitats de malalties minoritàries es recomana que hi hagi un psicòleg per abordar aquests aspectes. Ja que no pots curar, almenys intentem alleugerir la càrrega fent-los saber que hi ha un lloc on poden ser atesos de la seva malaltia, un lloc on s’investiga… Això també els millora
la qualitat de vida.
6. Edu

5 anys
Avinyonet del Penedès
SENSE DIAGNÒSTIC
L’Edu té cinc anys i viu a Avinyonet del Penedès amb la seva mare i la seva germana. Ell pertany al grup, encara més minoritari, dels malalts sense diagnòstic, i la seva mare, la Sandra Iglesias, presideix l’Associació Objectivo Diagnóstico, que agrupa trenta famílies de malalts sense diagnosticar d’arreu de l’Estat espanyol.
L’Edu mai ha pogut obrir els ulls per ell mateix, l’han operat tres vegades de les parpelles i està pendent de tornar a entrar a la sala d’operacions perquè les que li han fet fins ara no han funcionat com s’esperava. Això li provoca problemes de visió severs. A més té altres dolences: quan tenia cinc mesos el van operar d’una hèrnia diafragmàtica per controlar el reflux i té hipotonia, és a dir, el to muscular baix.
“El primer ingrés va arribar a la setmana de néixer i des de llavors ha estat un anar i venir”, comenta la mare. Durant els primers mesos de vida a l’Edu també van localitzar-li dos quists al cap que, per ara, no li provoquen cap disfunció.
A l’Edu el tracten metges de l’hospital de Sant Joan de Déu i des de fa un any està esperant que els investigadors aconsegueixin fons per fer-li un exoma trio, és a dir, una prova genètica molt exhaustiva d’ell i dels seus pares. “El pitjor de tot és esperar el diagnòstic. És desesperant, te’l mires amb lupa i estàs en alerta constant. Si li poguéssim posar nom a la malaltia, podríem saber si hi ha més afectats i compartir coneixement i experiència”, comenta la Sandra.
L’Edu va sempre que pot a l’escola d’Avinyonet del Penedès i aviat tindrà un professor de suport: “Fa mesos que ho estàvem esperant i fa pocs dies l’ONCE ens va comunicar que l’hi posaran”, comenta la seva mare. Per millorar el seu to muscular va a la piscina, fa teràpia amb cavalls i va a un centre d’arts marcials.
7. Pau

5 anys
Sant Sadurní d’Anoia
SÍNDROME DE MOWAT WILSON
I CORNELIA DE LANGE (en estudi)
El Pau va néixer a la setmana 30 d’embaràs amb només un quilo. La seva mare, la Gisela Coral, feia setmanes que estava ingressada a l’Hospital Sant Joan de Déu perquè a la setmana vint en veure que el fetus no creixia els metges van fer-li l’amniocentesi i la bossa es va trencar. Tot i que els resultats de la prova van sortir bé, el baix pes feia sospitar als metges que hi podia haver quelcom. Després de cinc mesos a l’hospital el Pau va arribar a casa acompanyat d’una màquina d’oxigen a la qual va estar connectat nit i dia fins que va tenir un any i mig. Aviat es van adonar que tenia una fissura al paladar, un tret sindromàtic que va fer que l’equip mèdic pensés en alguna malaltia minoritària.
“Fins que el Pau va tenir quatre anys no vam tenir els primers resultats de les proves genètiques”, explica la Gisela. Al Pau li van estudiar 6.710 gens associats i segons els resultats obtinguts pateix, com a mínim, dues malalties: Síndrome Mowat Wilson i Cornelia de Lange. “Tot i això, els metges ens han dit que el resultat és incert i que cal continuar estudiant, per això l’any vinent li repetiran la prova però ampliant a 8.000 gens”, explica.
El Pau madurativament avança molt lentament i en les seves ressonàncies cerebrals surten anomalies. Tot i que camina, té problemes motrius, té epilèpsia i presenta també trets autistes.
La Gisela assenyala com és de complicat el procés d’acceptació: “Al principi tot era incertesa, teníem por cada vegada que havíem d’anar al metge. Amb el temps, però ho vas assimilant i t’adones de tot el que aprens amb ell, tot el que ens aporta i com valores coses que abans ni et plantejaves”. Ara el Pau va unes hores a l’escola del Carme de Sant Sadurní i ho combina amb teràpies.
8. Lourdes Sors

58 anys
La Bleda (Sant Martí Sarroca)
POST-POLIOMELITIS
Quan la Lourdes tenia 18 mesos va contreure el virus de la poliomelitis “existia la vacuna i en diversos països europeus es posava, però a l’Espanya Franquista s’havia de pagar i moltes famílies no s’ho podien permetre”, explica.
El virus li va deixar coixesa permanent i diverses patologies associades, “però sempre he intentat portar una vida normal, he treballat des dels 14 anys fins que m’he pogut jubilar, he tingut un fill…”.
A la Lourdes la sorpresa li va arribar als 45 anys quan va començar a desencadenar diferents patologies: “queia molt sovint, perdia força i tenia molt dolor,”, comenta. Després de deu anys de pelegrinatge per metges i de patir dolor permanent, finalment la van derivar a l’Institut Guttmann on van diagnosticar-la de la síndrome de post-Poliomelitis. “El virus, que estava adormit des de fa anys, s’ha revifat, m’ataca les poques parts bones que em queden i me les malmet per sempre”, explica. La Lourdes està pendent d’una operació de malucs, serà la vint-i-tresena per ella, ja que al llarg de la seva vida l’han operat vint-i-dues vegades dels peus. Per fer més suportable el dolor es pren tota mena d’antiinflamatoris i pateix pels efectes secundaris que pugui tenir.
“La meva malaltia no té cura, estan investigant però no hi ha prou diners. A més, és una malaltia que morirà amb nosaltres i això tampoc ajuda”, denuncia. El que la Lourdes i molts malalts com ella reivindiquen és que siguin reconeguts com a víctimes del franquisme i que com a mínim se’ls subvencionin les teràpies que poden alleugerir-los el dolor.
9. Martina
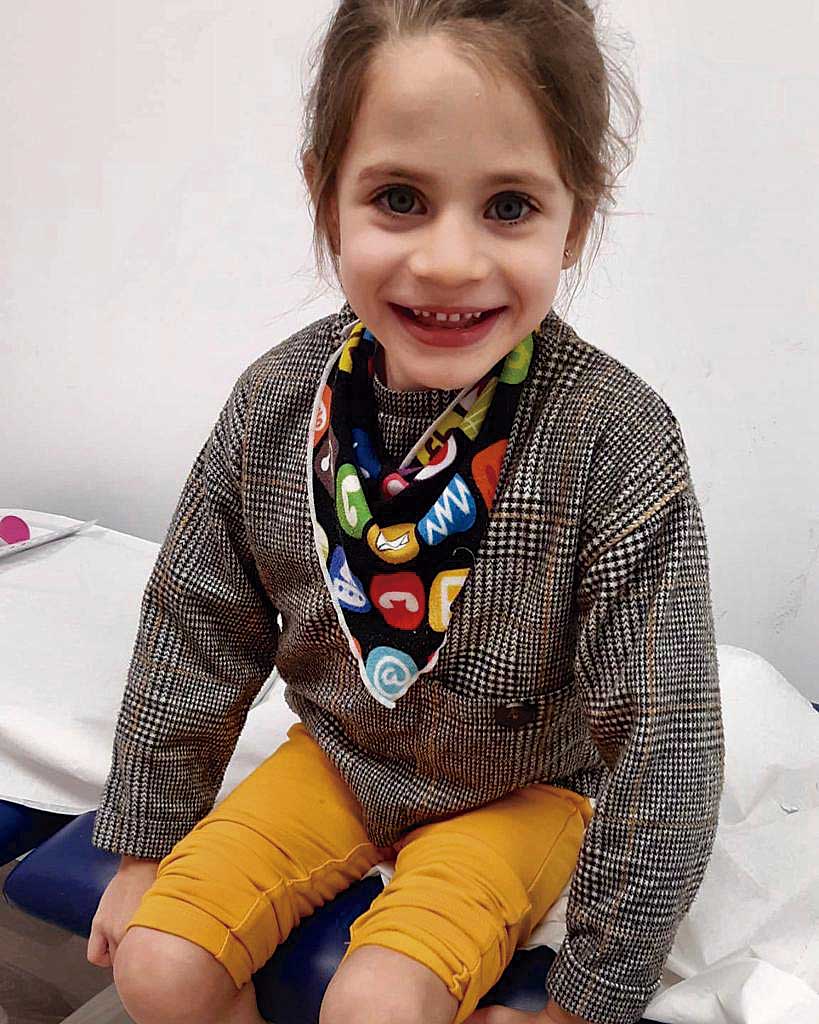
5 anys
Vilafranca del Penedès
HOMOCISTINURIA AMB ACIDÚRIA METILMALÒNICA (CBLC)
La Martina té cinc anys i una malaltia metabòlica rara anomenada Homocistinuria amb Acidúria Metilmalònica (CblC).
Després d’un embaràs i un part normal, la malaltia va debutar-li als quinze dies amb una crisi: “La Martina no sintetitza bé una proteïna i la transforma en tòxica. Nosaltres vam rebre la prova del taló tard i per correu ordinari, ningú abans ens va advertir que havia sortit malament, i hores després de rebre l’informe la Martina va tenir convulsions. Feia quinze dies que li donàvem llet i ella no en pot prendre, s’estava intoxicant i els òrgans li començaven a fallar”, comenta la Maria Parés, la seva mare. A Sant Joan de Déu va estar 48 hores en observació fins que una nova crisi la va deixar en estat molt crític. Va entrar a la UCI amb una fallada multiorgànica que li va provocar la discapacitat psíquica que pateix i que no està associada a la malaltia. A més, té una lesió a la màcula dels ulls que fa que no hi vegi bé.
Diàriament la Martina ha de prendre cinc pastilles diferents i li han de posar una injecció de vitamina B12. A més, durant els dos primers anys de vida va haver de portar una alimentació molt restrictiva sense ingerir pràcticament cap proteïna. A través de les xarxes socials la Maria i el Toni, el pare de la Martina, van conèixer el cas d’una nena italiana amb la mateixa malaltia que menjava de tot: “els hi vam explicar als nostres metges i vam decidir fer el mateix. Des que menja normal les analítiques li surten molt bé”, expliquen.
La Martina fa P4 a l’escola Baltà Elias de Vilafranca i pràcticament cada tarda fa alguna teràpia. A més, els seus pares fa gairebé dos anys van crear l’associació #loverare per divulgar les malalties minoritàries. Justament, el mes d’octubre, per exemple, van organitzar un concert familiar a Vilafranca per recaptar fons per La Marató de TV3.
10. Immaculada Sánchez

63 anys
Vilafranca del Penedès
DISTRÒFIA MUSCULAR MIOTÒNICA DE STEINERT
La Immaculada va descobrir que tenia distròfia muscular miotònica quan va ser mare, tot i que la malaltia no li va debutar fins als 45 anys. El seu fill Marc pateix la mateixa patologia que ella i quan tenia cinc anys el van diagnosticar: “els metges em van dir que era una malaltia hereditària i així vaig saber que jo també la tenia”, explica. Tot i això, els primers símptomes van trigar vint anys més a aparèixer-li. Ara li costa respirar, té un to muscular molt baix, cada vegada té més dificultats per moure’s i té problemes de visió: “La nostra malaltia no té cura, però els efectes depenen molt dels dies, dilluns per exemple vaig sortir a comprar i vaig caure dues vegades”.
La Immaculada viu des de fa cinc anys a Vilafranca amb el seu fill Marc, de 40 anys, que també té una deficiència intel·lectual associada a la malaltia. Viuen sols en un pis i compten amb l’ajuda d’una persona per a les tasques de neteja personal i una altra per a la de la llar, a més d’un servei de càtering a domicili tots els migdies. Es tenen l’un a l’altre i, per ara, el Marc no va a cap centre: “Quan vivíem a l’Hospitalet sí que hi anava, però per arribar-hi havíem d’agafar cada dia el tren i l’autobús, si em trobés com ara ja no el podria acompanyar”, comenta.








{kind=link}